A research team led by Dr Chaogu ZHENG from the School of Biological Sciences at The University of Hong Kong (HKU) has recently made a significant discovery about the evolutionary age of different type of cells in a small animal called Caenorhabditis elegans (C. elegans). By using single-cell transcriptomic data and refined phylostratigraphy, the team determines the transcriptomic age of individual cells, which means they are able to estimate the evolutionary origin of different cells based on the age of the genes expressed in the cells.
Their findings shed light on the cellular basis of the ‘hourglass’ pattern of animal development, revealing significant variation in the transcriptome age of different cell types. These results also provide insights into the varying contribution of different cells and tissues to adaptation, and the evolutionary relationship among cell types. These findings offer new perspectives on the genetic mechanisms that drive the evolution of species and have been published in the leading multidisciplinary journal PNAS.
Insights from Molecular Studies on Hourglass Model
The embryos of all animals share similar morphology at the middle stage of embryonic development while having larger morphological divergence at earlier and later stages. This pattern is often referred to as the ‘hourglass’ pattern of development, meaning that all animal development experiences an evolutionarily conserved phase during mid-embryogenesis.
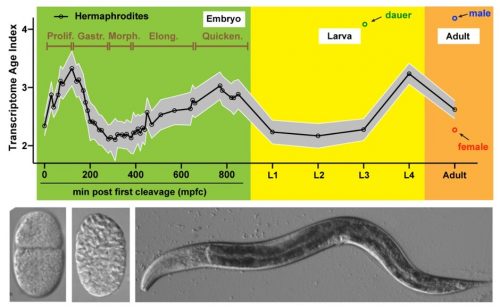
Recent molecular studies have shown that embryos at the middle stage of embryogenesis express the oldest transcriptome, which means that the oldest and most conserved genes are used at this stage during gene expression. In contrast, younger genes are expressed in the earlier and later stages of embryonic development. This was discovered by analysing gene expression of the embryos in different developmental stages using a technique called phylostratigraphy, a method used to determine gene ages by comparing their sequences across different species.
However, these studies are limited in that they could only determine the transcriptome age of the entire organism throughout development but not in individual cells or tissue. This limitation is significant because obtaining information about the age of genes expressed in specific cells and tissue is crucial for gaining a more detailed understanding of the evolution of developmental patterns among species, as well as the genetic mechanisms driving it. Additionally, it can shed light on how individual tissue and cells contribute to the ‘hourglass’ pattern, which is a crucial aspect of understanding how different organs and tissues contribute to the evolution and adaptation of the overall developmental process in animals.
From Whole-Organism to Single-Cell Analysis
To fill this knowledge gap, the research team studies the transcriptome age of the nematode C. elegans at the single cell level using RNA sequencing. They look at RNA expression from both whole embryos (or organism) and individual cells to gain a comprehensive understanding of how different genes are used during embryonic and larval development.
The team first identifies a period of the oldest transcriptome during C. elegans mid-embryogenesis, which starts after gastrulation, a process that forms different germ layers in the embryo and continues into the early development of an organ. More importantly, the research team finds that in early embryos, certain cells used older genes than other cells. For example, cells that would later become the germline (which is responsible for passing on genetic information to offspring) use older genes than somatic tissues in the body. Similarly, cells that would later become the endoderm (which gives rise to the digestive tract) use older genes compared to other cell types during early development. Among differentiated cells, muscles appear to have the oldest transcriptome than other cell types.
It is also observed that the variation in transcriptome ages among the cell and tissue types remain small in early embryonic stages and grow bigger at late embryonic and larval stages as cells differentiate. Tracking the dynamics of transcriptome age along lineages identifies certain tissues, such as the skin, that contribute to the rise of the transcriptome age in late embryos.
Further analysis of the variation in transcriptome ages among the 128 different types of neurons in C. elegans nervous system reveals that a specific group of chemosensory neurons and their downstream interneurons express very young transcriptomes, which may have contributed to adaptation in recent evolution, as many newly evolved young genes are associated with sensing environmental factors. Finally, by analysing the variation in transcriptome age among the different neuron types, as well as the age of the genes that regulate their development (fate regulators), the research team is able to hypothesize about the evolutionary history of some of these 128 neuron types.
‘Using C. elegans as an example, we showcase how the transcriptome age at the single-cell level can provide insight into the cellular basis of developmental innovation and help understand the functional diversity and evolutionary origin of cell types,’ said Dr Fuqiang MA, a Postdoctoral Fellow of HKU School of Biological Sciences and the first author of the paper.
Dr Zheng, the supervisor of the research project, highlighted that ‘this study serves as an example of using the cutting-edge single-cell transcriptomics to study old problems in evolutionary biology.’ Dr Zheng envisions that the possibility of determining the evolutionary age of individual cell types at the transcriptome level can open up new research directions and advance our understanding of the genetic mechanisms that drive the evolution of species.
About the research paper: Ma F, Zheng C. Transcriptome age of individual cell types in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2023 Feb 28;120(9):e2216351120. doi: 10.1073/pnas.2216351120.
The journal paper can be accessed from here
This work is supported by funding from the National Science Foundation of China, the Research Grant Council of Hong Kong, and The University of Hong Kong.